AMILOIDOSIS CARDÍACA
Es una enfermedad causada por el depósito de proteínas anormales (amiloide) en los tejidos. El depósito suele ser progresivo y, dependiendo del órgano afectado, su función puede verse alterada.

El corazón se ve afectado en uno de cada tres o cuatro pacientes con amiloidosis.
Descripción
La amiloidosis cardiaca es una enfermedad progresiva y grave que se produce cuando es el corazón el órgano en el que se deposita el amiloide.
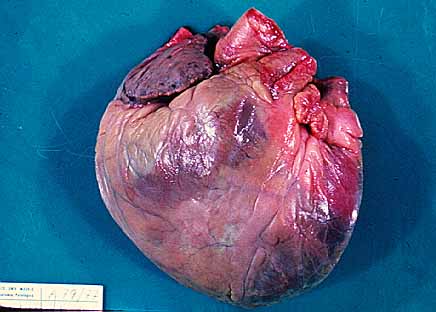
Es más frecuente en hombres que en mujeres y es excepcional antes de los 40 años.
Causas
En ocasiones la producción y depósito del material amiloide se relaciona con la presencia de ciertas enfermedades (mieloma múltiple, artritis reumatoide…) o como consecuencia de la hemodiálisis a largo plazo.

No obstante también existen amiloidosis familiares de causa genética y una forma denominada senil que aparece en personas mayores de 60 años y cuya causa es desconocida.
Síntomas
Puede producir cuatro síndromes cardiovasculares solapados:
- Miocardiopatía restrictiva. Se caracteriza porque el corazón se vuelve muy rígido y tiene dificultades para llenarse. Clínicamente se traduce en insuficiencia cardiaca con fatigabilidad, intolerancia al esfuerzo y síntomas congestivos (hinchazón del vientre y de las zonas declives).
- Insuficiencia cardiaca congestiva sistólica. Caracterizada por un deterioro progresivo de la fuerza contráctil del corazón. También se traduce en fatigabilidad, sensación de falta de aire y síntomas congestivos. Además los pacientes pueden presentar también angina de pecho.
- Hipotensión ortostática. Producida por el depósito del material amiloide en los vasos sanguíneos y el sistema nervioso vegetativo. Puede producir mareos e incluso síncopes (pérdida de consciencia transitoria) relacionados con el esfuerzo físico o la tensión emocional.
- Trastornos del sistema de conducción. Pueden aparecer arritmias, tanto taquiarritmias (>100 latidos por minuto) como bradiarritmias (<60 latidos por minuto).
Diagnóstico
El diagnóstico se sospecha ante determinadas manifestaciones clínicas y con las pruebas cardiológicas habituales, como el electrocardiograma (que registra la actividad eléctrica del corazón), el ecocardiograma o la resonancia magnética cardiaca (que permiten ver sus estructuras).
El diagnóstico de certeza se obtiene con la biopsia, es decir, tomando una muestra de tejido del órgano afectado en la que se objetiva el depósito del material amiloide. Se puede hacer biopsia de la grasa abdominal, del recto, de las encías, de la médula ósea, del hígado o del riñón. La biopsia cardiaca da el diagnóstico de amiloidosis cardiaca, pero otras localizaciones también ofrecen resultados y resultan menos agresivas.
El diagnóstico de certeza se obtiene con la biopsia, es decir, tomando una muestra de tejido del órgano afectado en la que se objetiva el depósito del material amiloide. Se puede hacer biopsia de la grasa abdominal, del recto, de las encías, de la médula ósea, del hígado o del riñón. La biopsia cardiaca da el diagnóstico de amiloidosis cardiaca, pero otras localizaciones también ofrecen resultados y resultan menos agresivas.
Tratamiento
El tratamiento es el de la enfermedad causal, si la hubiera (quimioterapia si existe un mieloma, inmunosupresores en la artritis reumatoide, etc.) y el de las manifestaciones cardiacas que aparezcan, sobretodo el de la insuficiencia cardiaca.
Pronóstico
El pronóstico vital de la enfermedad es, en general, malo a pesar del tratamiento.